TLT: What is your scientific background and how long have you worked in the computational tribology field? How did you decide to study lubricants?
Righi: My scientific background is in computational condensed matter physics, focused on the study of surface and interface phenomena by first principles methods. These methods, also known as
ab initio methods, are based on solving the fundamental equations of quantum mechanics and allow for a highly accurate description of the atomistic interactions in materials because they explicitly consider the electrons, which govern the formation/rupture of chemical bonds.
I first met the tribology community in 2006 by participating in the Gordon Research Conference on Tribology held in Waterville, Maine, where the leading experts of the field were gathered. During the conference, I realized that the
ab initio methods, successfully applied at that time in many fields such as catalysis and surface physics/chemistry, were almost completely unknown by tribologists. This was rather surprising to me since tribology, and in particular tribochemistry, has much to do with surface chemistry and catalysis. I remember that it was there, at the Gordon Tribology Conference 2006, that I set myself the goal to port the
ab initio methods in the field of tribology, as I had the clear intuition that an accurate modeling of tribochemical reactions could bring great advantages to the development of new lubricant materials and additives.
TLT: How can computational methods help us understand the lubrication mechanism?
Righi: The macroscale friction coefficient is mainly governed by the adhesion of nano-asperity contacts. Indeed, the basic function of lubricants is to reduce that adhesion. In boundary lubrication conditions, this is accomplished either by covering the surfaces with inert films, such as diamond like carbon (DLC) and layered materials, or by forming lubricious films
in situ thanks to tribochemical reactions involving lubricant additives. To design effective lubricant additives is, thus, highly desirable to understand how tribofilms are formed and function. However, monitoring in real time the chemical reactions occurring at the buried sliding interface is nowadays impossible by experiments. Typically, by postmortem analysis of the wear tracks one can gain information of the reaction products, but very little can be inferred on the reaction paths and mechanisms. Atomistic simulations play a crucial role here as they allow us to open a window on the complex scenario of tribochemical processes and “observe” what happens during sliding. Moreover, simulations based on a quantum mechanical approach can provide fundamental understanding on the effects of molecular confinement and mechanical stresses on reaction kinetics. Such understanding is highly relevant for many research fields, such as mechanochemistry, where the use of mechanical forces to activate reactions is regarded as a unique opportunity to synthesize compounds with a reduced use of solvents.
TLT: What has been your most rewarding accomplishment throughout your career in tribology research?
Righi: My most rewarding accomplishments are related to the pioneering use of computational methods that I ported from the community of chemical physics to tribology, as detailed in the following.
In 2011, by implementing ad hoc modifications to a software for
ab initio molecular dynamics (AIMD), we were able to perform the first AIMD simulations of sliding interfaces, which provided novel understanding on the effects of humidity on the tribological properties of carbon-based films
(see Figure 1).1 I presented the work at the International Tribology Conference (ITC) in Hiroshima, Japan, attracting the interest of the scientific community as well as of the industries. Indeed, after the conference I was contacted by Toyota’s central R&D labs, and we started a collaboration for applying AIMD to design DLC coatings used in the car engine. I also was involved in a collaboration with TotalEnergies SA to design new, environmentally friendly lubricant additives.
2-6 Moreover, I started collaborating with leading experimental experts and used AIMD to provide insights into the tribochemistry and tribocatalysis processes that rule the measured frictional behavior, especially of 2D materials used as solid lubricants.
7-9
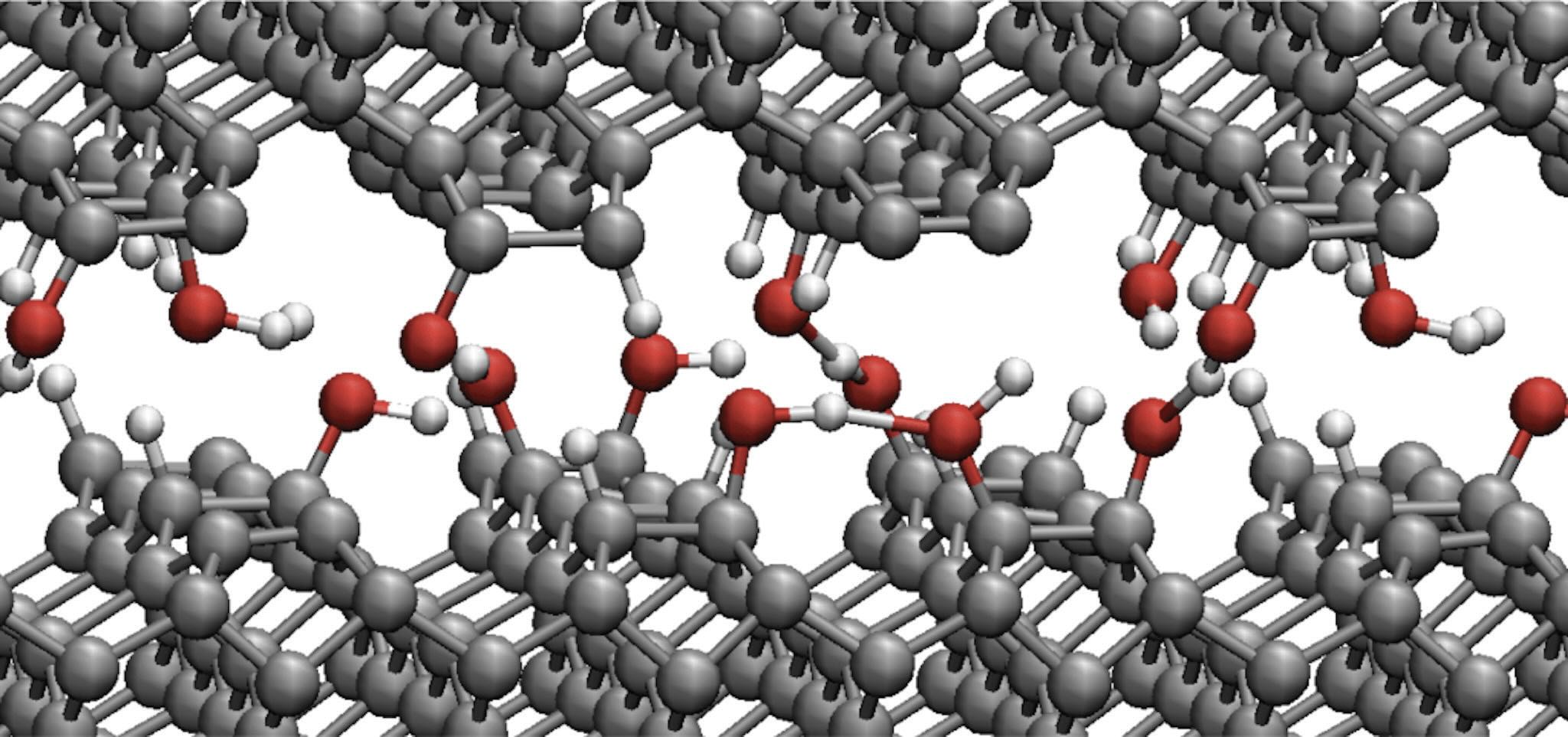 Figure 1. Snapshot acquired during the AIMD simulation of tribochemical reactions of water molecules at diamond interface.
Figure 1. Snapshot acquired during the AIMD simulation of tribochemical reactions of water molecules at diamond interface.
In 2018 I ported the high throughput first principles approach in tribology. Software developed by my group allows for screening the adhesion and shear strength of hundreds of solid interfaces in parallel
(see Figure 2).10 The populated databases permit us to uncover general trends.
11,12 For example, I discovered a relation between the electronic and tribological properties of interfaces, which sheds light into the fundamental origin of the friction force.
13 We are now using the software to design ad hoc surface chemical modifications to strengthen or weaken the interfacial adhesion of any material pairs.
14

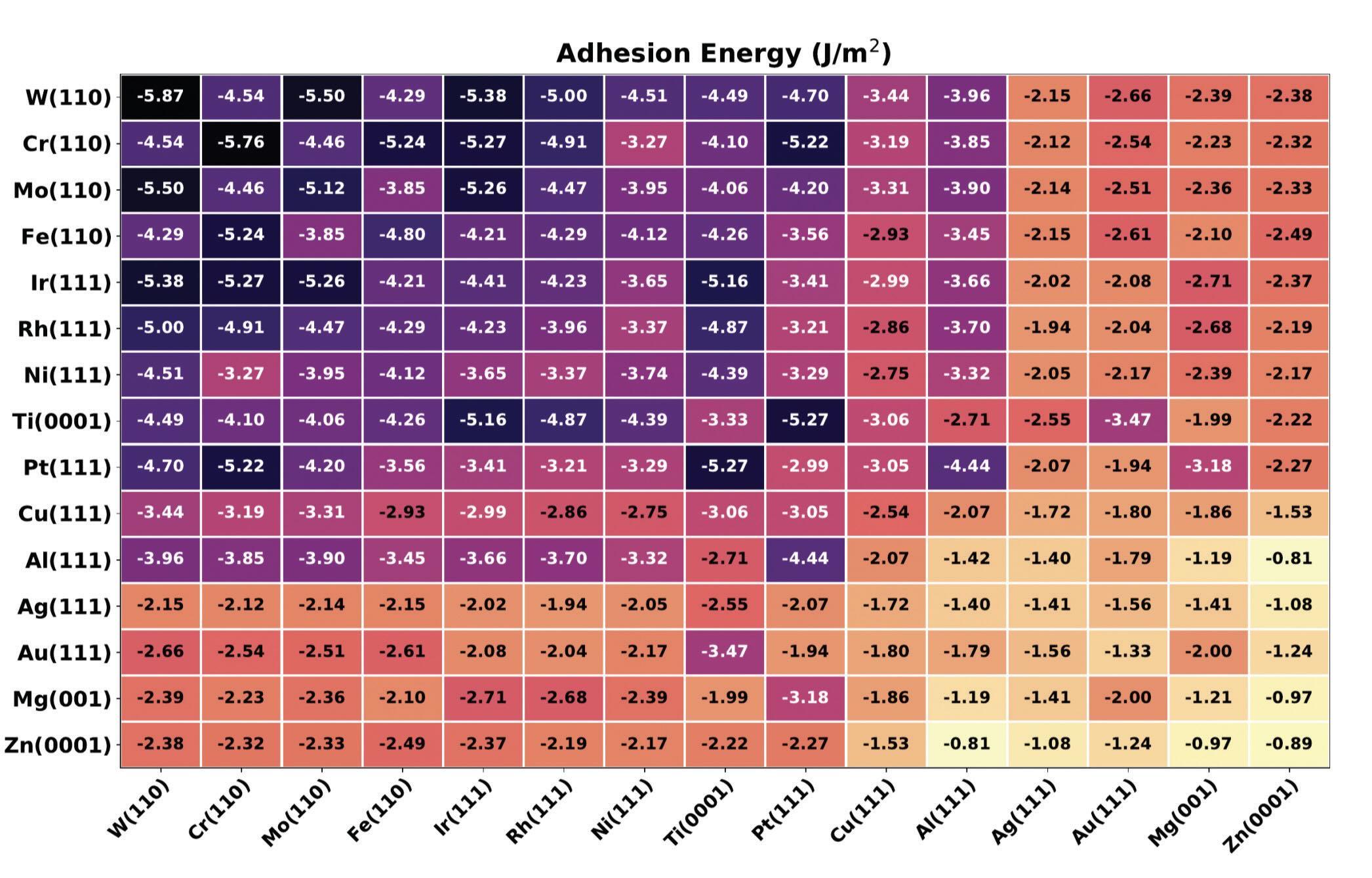 Figure 2. Schematic representation of the workflow implemented in the TRIBCHEM software for high throughput screening of solid interfaces (bottom). Database of first principles adhesion energies of metal-on-metal interfaces (top).
Figure 2. Schematic representation of the workflow implemented in the TRIBCHEM software for high throughput screening of solid interfaces (bottom). Database of first principles adhesion energies of metal-on-metal interfaces (top).
Last year we succeeded, after more than a one-year long period of struggling, in applying machine learning (ML) to accelerate molecular dynamics (MD) simulations of a large-scale tribological system. I presented the results, concerning lubricant additives confined at the iron interfaces
(see Figure 3),15 during the Tribochemistry Beppu 2023 Conference in Beppu, Japan, and it appeared evident that ML has the power to revolutionize the field of computational tribology as ML-MD simulations can offer the accuracy of
ab initio MD at the computational cost of classical MD. Therefore, it will be soon possible to simulate large systems, such as lubricant additives included in the liquid media, or multi-asperity interfaces in a very realistic way.
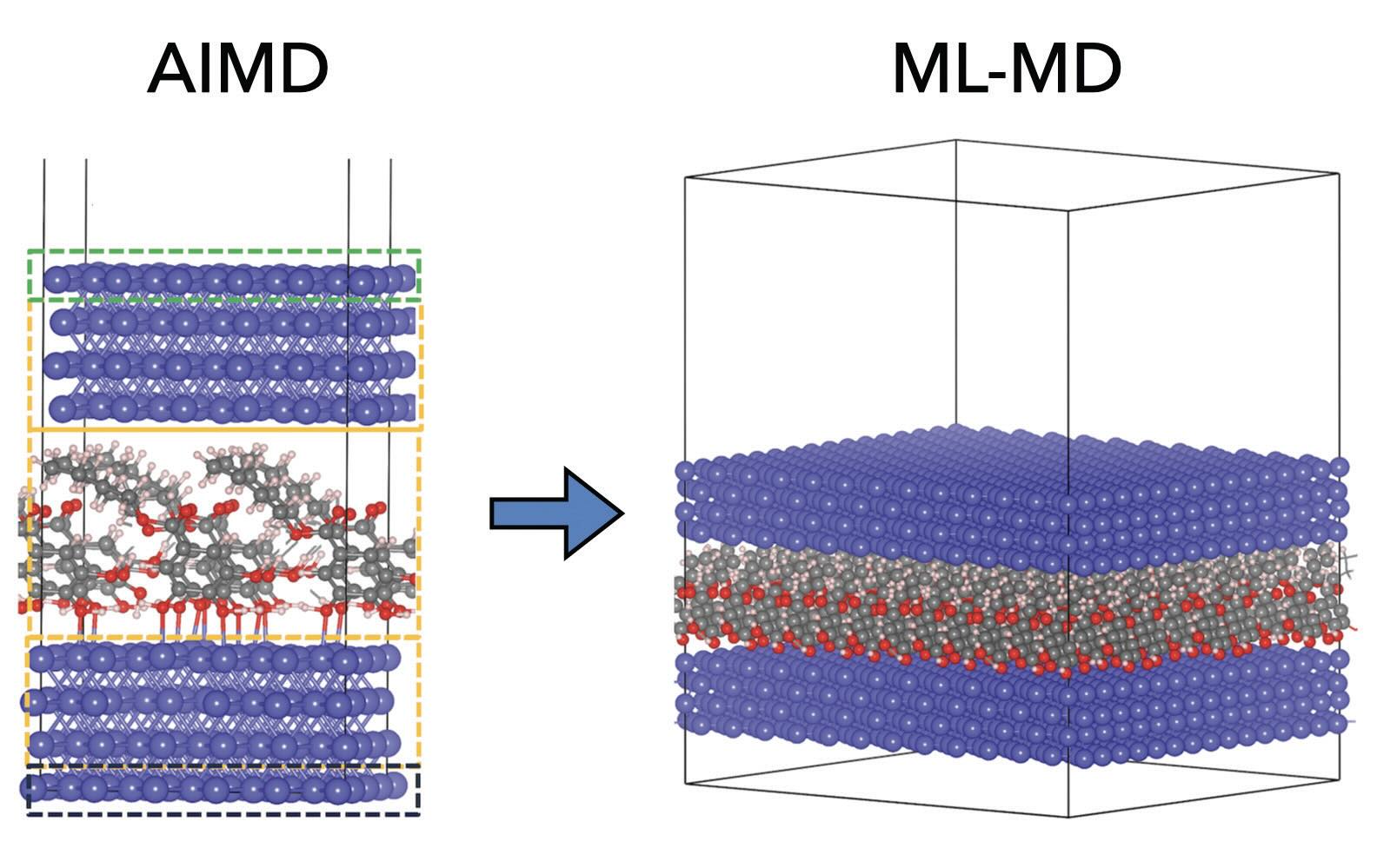 Figure 3. Lubricant molecules at sliding iron interfaces simulated by machine learning molecular dynamics (right) at almost the same accuracy and much lower computational cost that the AIMD simulations used for training (left).
Figure 3. Lubricant molecules at sliding iron interfaces simulated by machine learning molecular dynamics (right) at almost the same accuracy and much lower computational cost that the AIMD simulations used for training (left).
TLT: Throughout the different segments within your career, which one has been the most interesting, challenging and/ or rewarding?
Righi: The most rewarding and challenging step in my career is related to the grant of about 1.5 million euros that I received from the European Research Council (ERC) for the five-year project “Advancing Solid Interfaces and Lubricants by First Principles Materials Design” (SLIDE). The grant, awarded in 2019 based on excellence criteria, allowed me to be promoted as full professor and establish a group of young scientists at the University of Bologna, Italy, committed in advancing the computational tribology field by developing new computational methods and applying them to design new lubricant materials.
TLT: What are some of the most technical lubrication-based concepts or topics you have encountered throughout your career?
Righi: A key technical concept for friction reduction, which rules the functionality of per- and polyfluoroalkyl substances (PFAS) and of many other lubricants, including 2D materials, is the Pauli repulsion. This interaction, of quantum mechanical origin, makes the atoms with filled electronic shells behave as rigid spheres, like billiard balls, that interact through big repulsive force when they come into contact because they cannot interpenetrate each other. For example, when the fluorine atoms bind to the carbon atoms in the polytetrafluoroethylene (abbreviated as PTFE), they complete the electronic shells and repell any other incoming atom that may overlap its electronic density with that surrounding the polymer, which results completely inert. This mechanism explains why PTFE coatings can highly reduce the substrate adhesion with any countersurface making it slippery and hydrophobic.
16 The same concept explains the reason why layered materials, such as graphene and MoS
2, are good solid lubricants: thanks to the intralayer bonds the atoms constituting the layer fill their electronic shells. Consequently, a high Pauli repulsion arises between the layers at short range,
17,18 making the interlayer distances too high for any chemical interaction, and only weak van der Waals attraction survives.
A second, fascinating concept that I encountered in studying tribochemical processes concerns the effects of mechanical forces in activating chemical reactions. Our AIMD simulations, in agreement with the experimental observations, reveal that the reaction rates at the sliding interface are much higher than those of thermally activated processes occurring at the open surface. Indeed, during the short time intervals of our AIMD simulations we were able to observe the activation of complex processes such as the synthesis of new materials, e.g., graphene from methene molecules,
8 and MoSe
2 from selenium nanopowder,
7 as well as structural phase transitions leading to the crystallization of an amorphous materials.
9 Such highly energetically costly processes could occur in short time intervals thanks to the presence of the load and shear stresses that promoted the molecular confinement and diffusion, respectively. Predicting how these effects impact the reaction rates is key for harnessing mechanochemical reactions and use them in several fields to create new, useful compounds or destroy the harmful ones in a more efficient way than reactions activated by temperature or light.
TLT: Can simulations currently lead to the robust, rational design of effective lubricants?
Righi: My answer is definitely yes. I believe that the advanced simulations techniques nowadays available thanks to the knowledge transfer through different scientific communities have the power to greatly advance tribology. Integrating computational tools and digital data with experiments is highly beneficial for 1.) understanding on the functionality of lubricant materials and 2.) discovering new compounds with optimal lubricating properties.
1.) It is well accepted that MD simulations are very important to uncover the atomistic mechanisms that rule the frictional behavior of materials. In particular AIMD allows for an accurate description of the tribochemical processes that govern the functionality of many solid lubricants and lubricant additives in boundary lubrication conditions. With the advent of ML potentials, it has become possible to highly increase the size of the simulated systems and the duration of simulated time intervals without losing the accuracy of AIMD. This will greatly advance the research in lubricants, making it possible to realize highly realistic
in silico experiments and reduce the time and costs of lubricant deployment.
2.) High throughput first principles calculations have been successfully used to design new materials for many applications, from hydrogen storage to batteries. Their use in tribology is rather recent, but the preliminary results are encouraging. For example they allowed to create databases for the adhesion and shear strength of many materials pairs,
11 identify the fundamental origin of the adhesive friction
13 and design chemical surface modifications to decrease (or increase) it.
14 By adopting a high throughput approach in tribology experiments, large databases can be created and analyzed through ML techniques, thus extending the newly emerging field of “materials informatics” to tribology.
You can reach M. Clelia Righi at clelia.righi@unibo.it.
REFERENCES
1.
Zilibotti, G., Corni, S. and Righi, M.C. (2013), “Load-induced confinement activates diamond lubrication by water,”
Physical Review Letters, 111, 146101.
2.
Peeters, S., Charrin, C., Duron, I., Loehlé, S., Thiebaut, B. and Righi, M.C. (2021), “Importance of the catalytic effect of the substrate in the functionality of lubricant additives: the case of molybdenum dithiocarbamates,”
Materials Today Chemistry, 21, 100487.
3.
Peeters, S., Restuccia, P., Loehlé, S., Thiebaut, B. and Righi, M.C. (2020), “Tribochemical reactions of MoDTC lubricant additives with iron by quantum mechanics / molecular mechanics simulations,”
Journal of Physical Chemistry C, 124, 13688.
4.
Loehlé, S. and Righi, M.C. (2018), “Ab initio molecular dynamics simulation of tribochemical reactions involving phosphorus additives at sliding iron interfaces,”
Lubricants, 6, 31.
5.
Righi, M.C., Loehle, S., de Barros Bouchet, M.I., Mambingo-Doumbe, S. and Martin, J.M. (2016), “A comparative study on the functionality of S- and P-based lubricant additives by combined first principles and experimental analysis,”
RSC Advances, 6, 47753.
6.
Righi, M.C. Loehle, S., de Barros Bouchet, M.I., Philippon, D. and Martin, J.M. (2015), “Trimethylphosphite dissociative adsorption on iron by combined first-principle calculations and XPS experiments,”
RSC Advances, 5, 101162.
7.
Grützmacher, P.G., Cutini, M., Marquis, E., Rodríguez Ripoll, M., Riedl, H., Kutrowatz, P., Bug, S., Hsu, C.J., Bernardi, J., Gachot, C., Erdemir, A. and Righi, M.C. (2023), “Se nano-powder conversion into lubricious 2D selenide layers by tribochemical reactions,”
Advanced Materials, 35, 2302076.
8.
Ramirez, G., Eryilmaz, O.L., Fatti, G., Righi, M.C., Wen, J. and Erdemir, A. (2020), “Tribochemical conversion of methane to graphene and other carbon nanostructures: Implications for friction and wear,”
ACS Applied Nano Materials, 3, 8060.
9.
Peeters, S., Losi, G., Restuccia, P. and Righi, M.C. (2022), “Unravelling the mechanism to form MoS2 lubricant layers from MoDTC by ab initio simulations,”
Applied Surface Science, 606, 154880.
10.
Losi, G., Chehaimi, O. and Righi, M.C. (2023), “TribChem: a software for the first-principles, high-throughput study of solid interfaces and their tribological properties,”
Journal of Chemical Theory and Computation, 19, 5231.
11.
Restuccia, P., Losi, G., Chehami, O., Marsili, M. andnRighi, M.C. (2023), “High throughput first principles prediction of interfacial adhesion energies in metal-on-metal contacts,”
ACS Applied Materials & Interfaces, 15, 19624.
12.
Wolloch, M., Losi, G., Ferrario, M. and Righi, M.C. (2019), “High-throughput screening of the static friction and ideal cleavage strength of solid interfaces,”
Scientific Reports, 9, 17062.
13.
Wolloch, M., Levita, G., Restuccia, P. and Righi, M.C. (2018), “Interfacial charge density and its connection to adhesion and frictional forces,”
Physical Review Letters, 121, 026804.
14.
Poli, E., Cutini, M., Nosir, M.A.M.A., Chehaimi, O. and Righi, M.C. (2024), “Effects of surface chemical modifications on the adhesion of metallic interfaces. An high-throughput analysis,” submitted (arXiv version available).
15.
Ta, H.T.T., Ferrario, M., Loehlé, S. and Righi, M.C. (2024), “Ab initio informed machine learning potential for tribochemistry and mechanochemistry: Application for eco–friendly gallate lubricant additive,” submitted.
16.
Pacini, A., Ferrario, M., Loehlé, S. and Righi, M.C. (2024), “Advancing tribological simulations of carbon-based lubricants with active learning and machine learning molecular dynamics,” submitted.
17.
Damiani, E., Marsili, M. and Righi, M.C. (2024), “Pauli repulsion at the heart of the amazing lubricating properties of PFAS and their possible substitutes,” submitted.
18.
Losi, G., Cutini, M., Restuccia, P. and Righi, M.C. (2023), “Modeling phosphorene and MoS2 interacting with iron: lubricating effects compared to graphene,”
Journal of Nanostructure in Chemistry, 13, 497.